


Cancer precursor project - breast cancer, part 6m
26 May 2025
This weekly medical blog is dedicated to science, not politics. For political commentary, I maintain a separate daily blog. You're welcome to skip any essays or sections that don’t interest you - no offense taken.
This post is part of my ongoing cancer precursor project, which explores how various cancers develop. Part 6 focuses on breast cancer. You can click here for links to essays on all 45 types of breast cancer (some are still in progress).
In this essay, I examine four more examples of triple negative breast cancer: angiosarcoma including postradiation, epithelioid angiosarcoma, breast implant associated anaplastic large cell lymphoma and myoepithelial carcinoma. These malignancies originate from cells that are triple negative, consistent with their non epithelial differentiation.
Angiosarcoma of the breast
Angiosarcoma of the breast is exceptionally rare (< 0.04% of all breast cancers). It arises from the endothelial cells lining blood vessels or lymphatics in the breast. It is classified as either primary angiosarcoma, which occurs de novo or secondary angiosarcoma, which typically develops following radiation therapy, chronic lymphedema or both.
Primary breast angiosarcoma predominantly affects younger women with a median age of 35 years. It often presents as a rapidly enlarging, painless mass, sometimes accompanied by bluish skin discoloration. Unlike secondary angiosarcoma, it usually develops within the breast, not in the skin or dermal layers. Diagnosis is challenging due to its rarity and nonspecific imaging features. Mammography may be inconclusive, especially in dense breast tissue, but MRI is more sensitive. Core needle biopsy is preferred over fine needle aspiration to identify malignant blood vessels.
Secondary angiosarcoma typically occurs in older women (median age 70 years). It arises in previously irradiated breast tissue, usually 7 to 10 years after radiation therapy or due to chronic lymphedema (Stewart-Treves syndrome). Clinically, it often presents with skin changes in the irradiated area, including purplish discoloration or nodules.
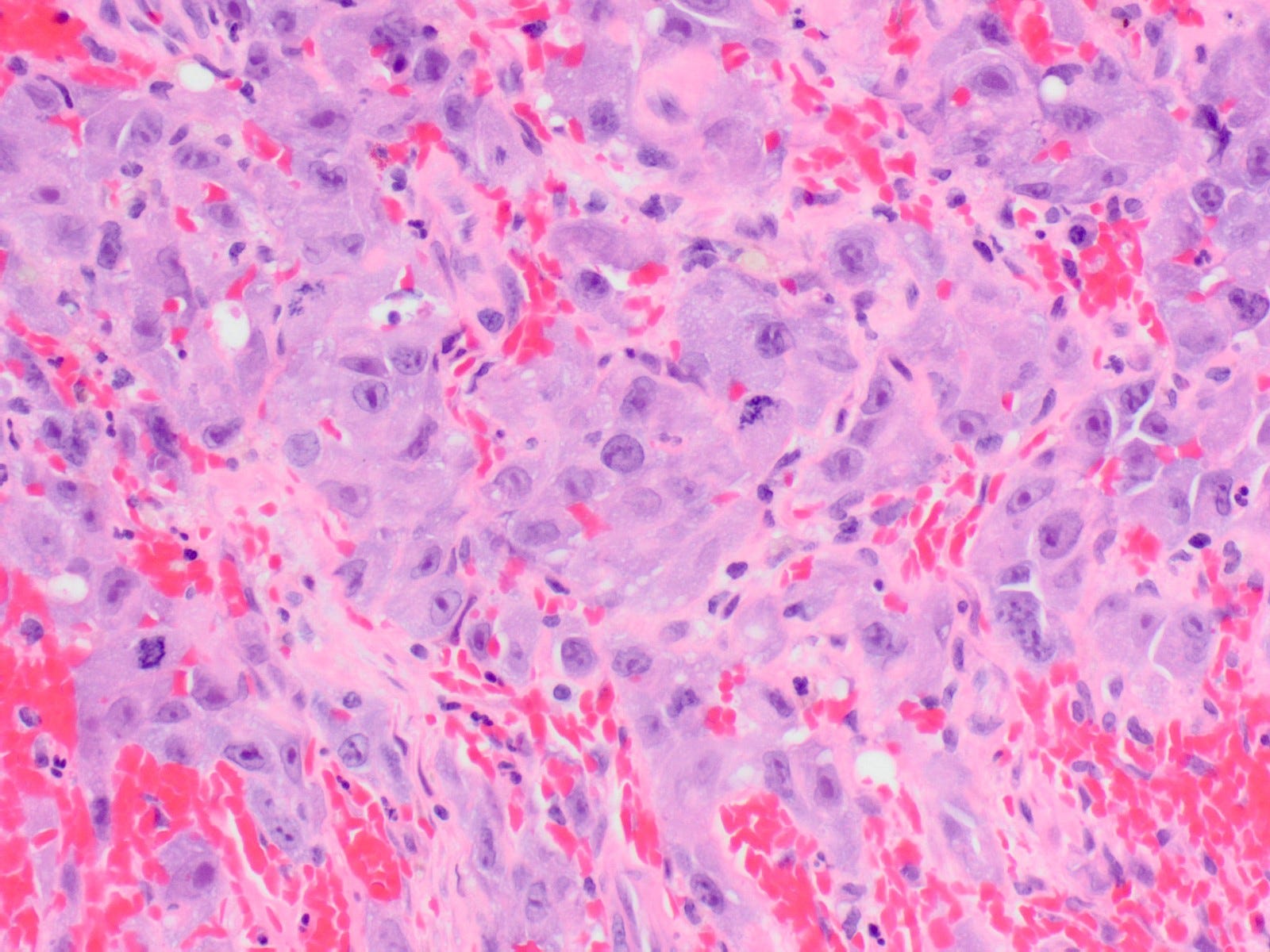
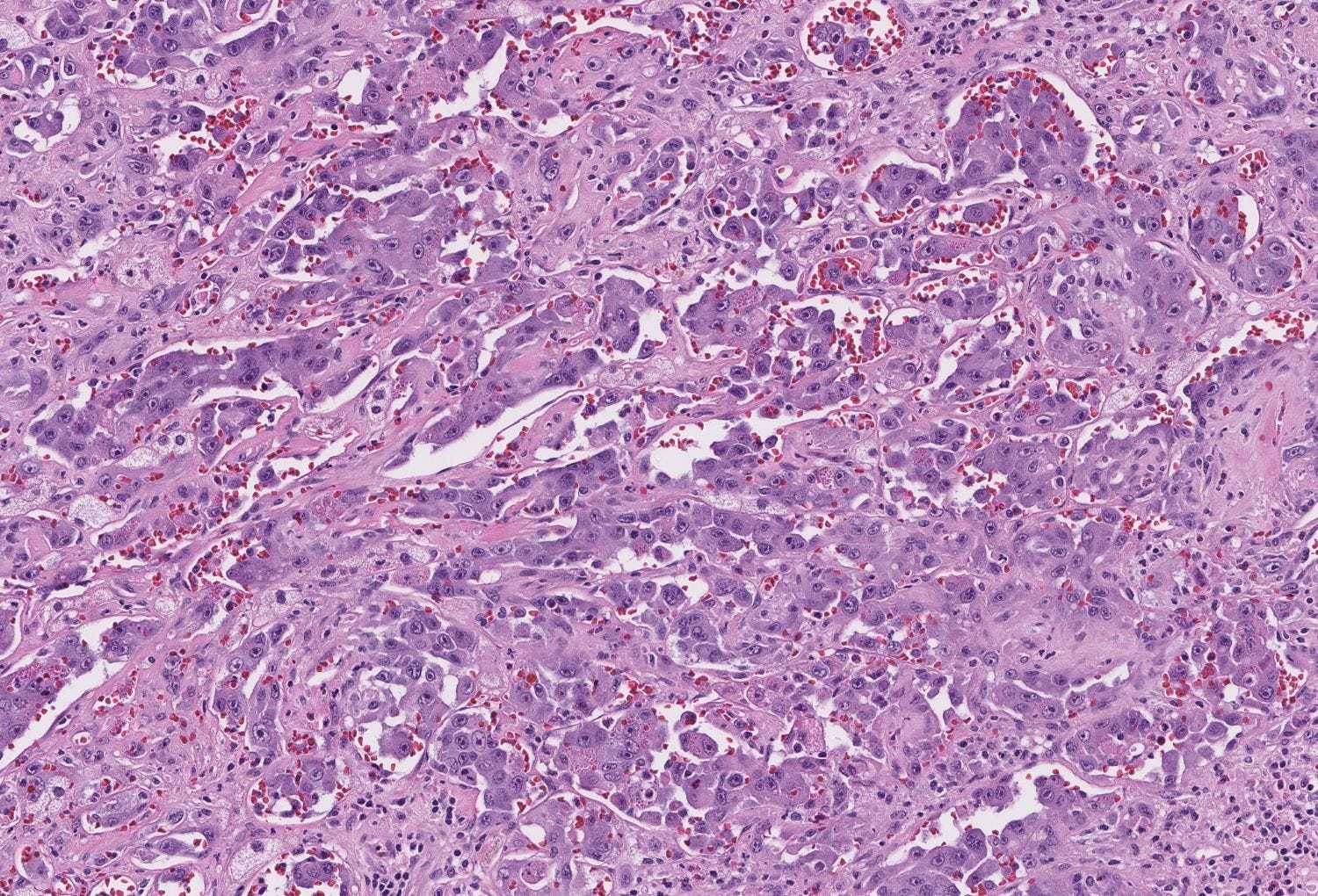
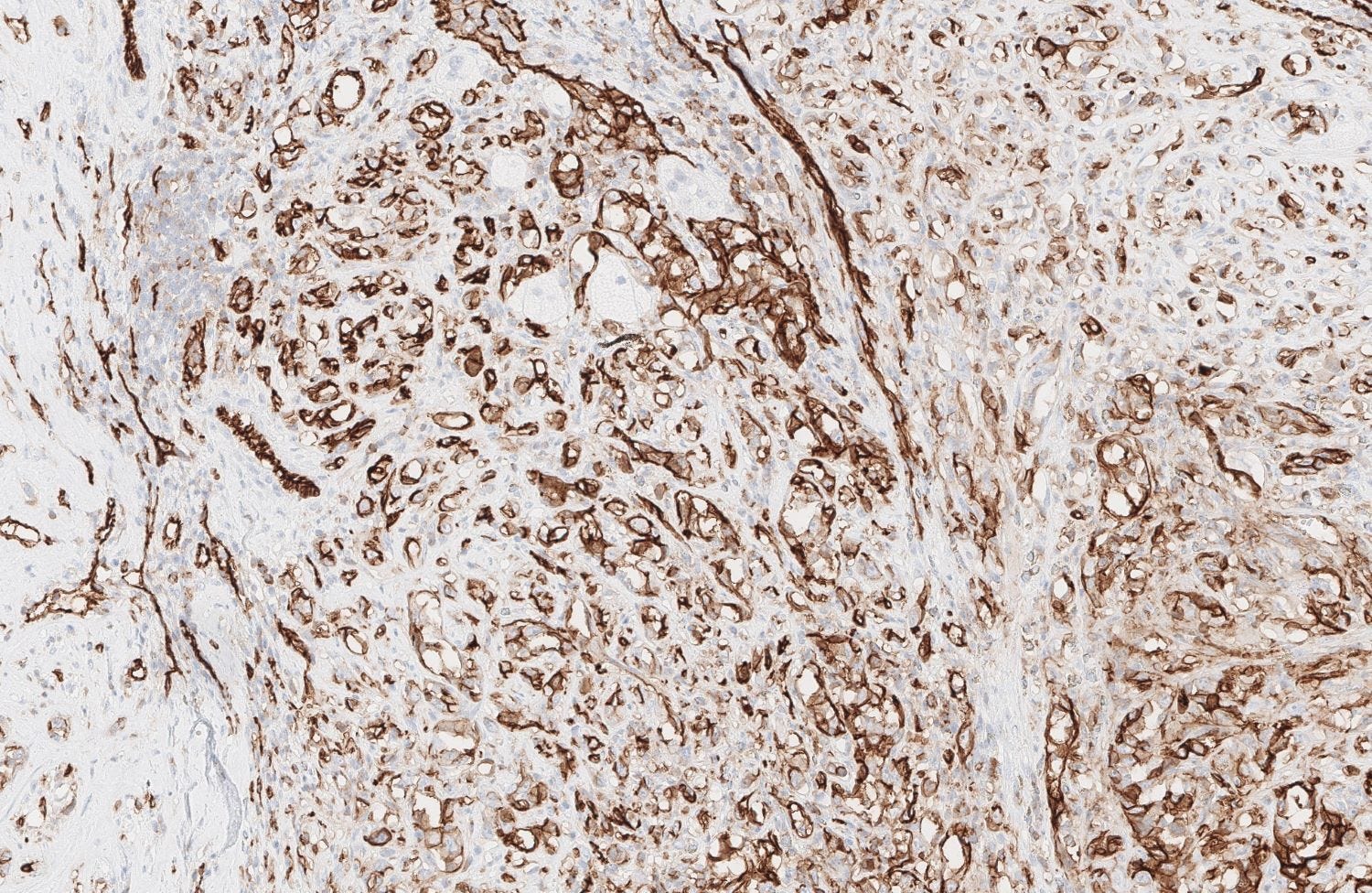
Histologically, both types of angiosarcoma display well formed vascular channels in low grade lesions. Higher grade lesions display solid sheets of atypical endothelial cells, epithelioid or spindle cells with marked cellular atypia and increased mitotic activity. Immunohistochemical staining is crucial for diagnosis: tumor cells express endothelial markers CD31, CD34, ERG and FLI1. Breast angiosarcomas appear to be triple negative (i.e. negative for ER, PR and HER2), consistent with their endothelial (non-epithelial) origin.
Treatment consists of surgical excision with negative margins, which may require wide excision in large tumors. Routine axillary lymph node dissection is generally not indicated because angiosarcoma primarily spreads via the bloodstream, not lymphatics. The effectiveness of chemotherapy and radiotherapy is unclear, with studies showing conflicting results.
Angiosarcoma is an aggressive tumor with a poor prognosis. Five year overall survival is only 25% to 40%, with high grade tumors and positive surgical margins being associated with increased risk of recurrence and decreased survival.
Angiosarcoma arises from the malignant transformation of endothelial cells or their progenitors, which may derive from mesenchymal stem cells. In general, no precursor lesion has been identified although atypical vascular lesions are rarely observed. Endothelial cell proliferation is normally triggered by pro-angiogenic signals that promote the formation of new blood vessels, a physiologic process essential during embryogenesis, wound healing, inflammation, the menstrual cycle, muscle growth and organ lining regeneration.
Under normal physiologic conditions, angiogenesis is tightly regulated by vascular endothelial growth factor (VEGF), which is released in response to hypoxia, injury or inflammation. These processes are self-limiting because the initiating stimuli are transient and feedback mechanisms suppress continued endothelial proliferation.
In contrast, radiation exposure and chronic lymphedema, the major risk factors for secondary angiosarcoma, are not self-limiting. Radiation exposure induces permanent DNA damage in endothelial cells and their precursors and may stimulate sustained VEGF expression. Chronic lymphedema produces persistent inflammation, fibrosis and tissue hypoxia, further driving abnormal and continuous vascular proliferation that may culminate in malignant transformation.
Breast angiosarcoma - radiologic, gross and microscopic images
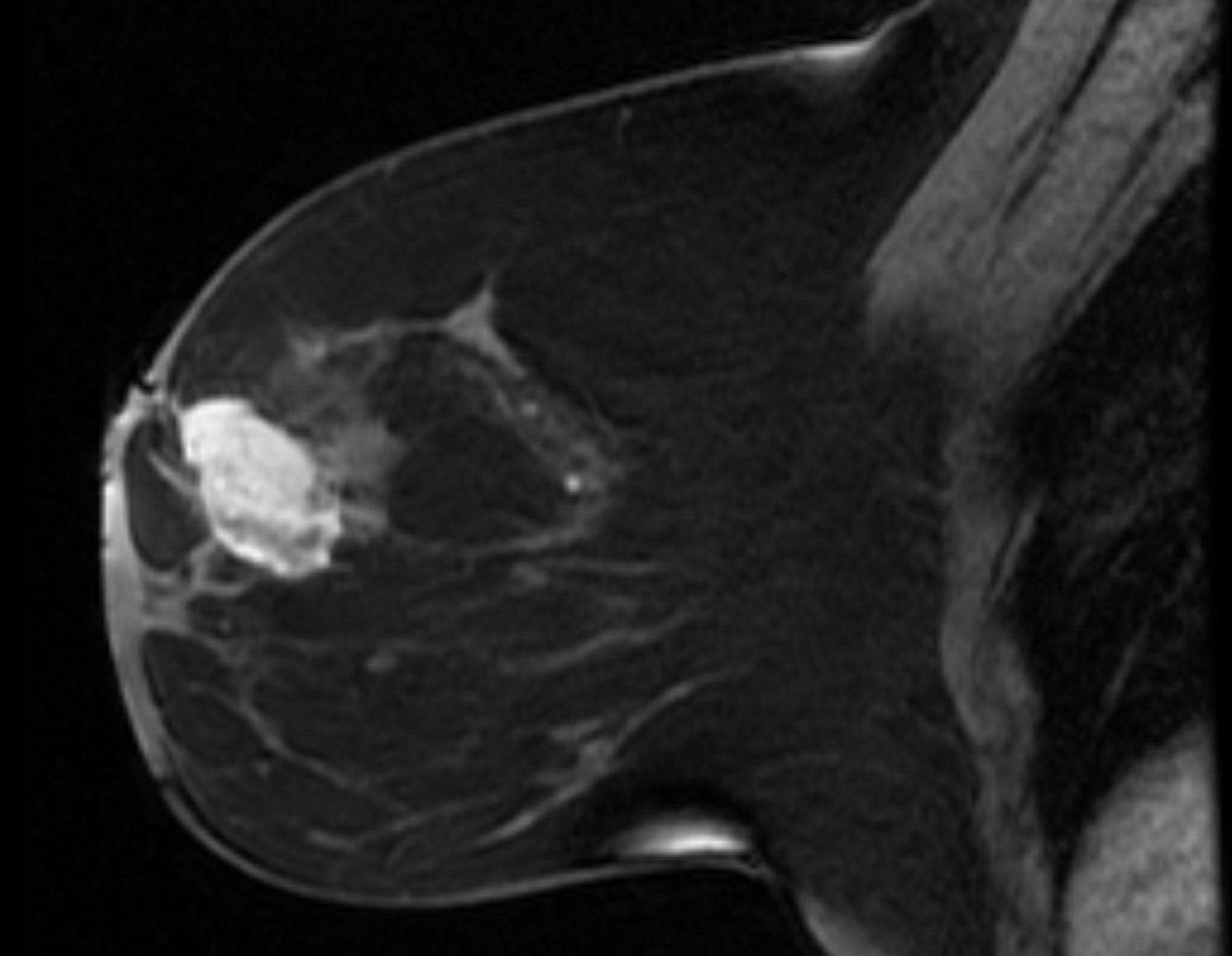
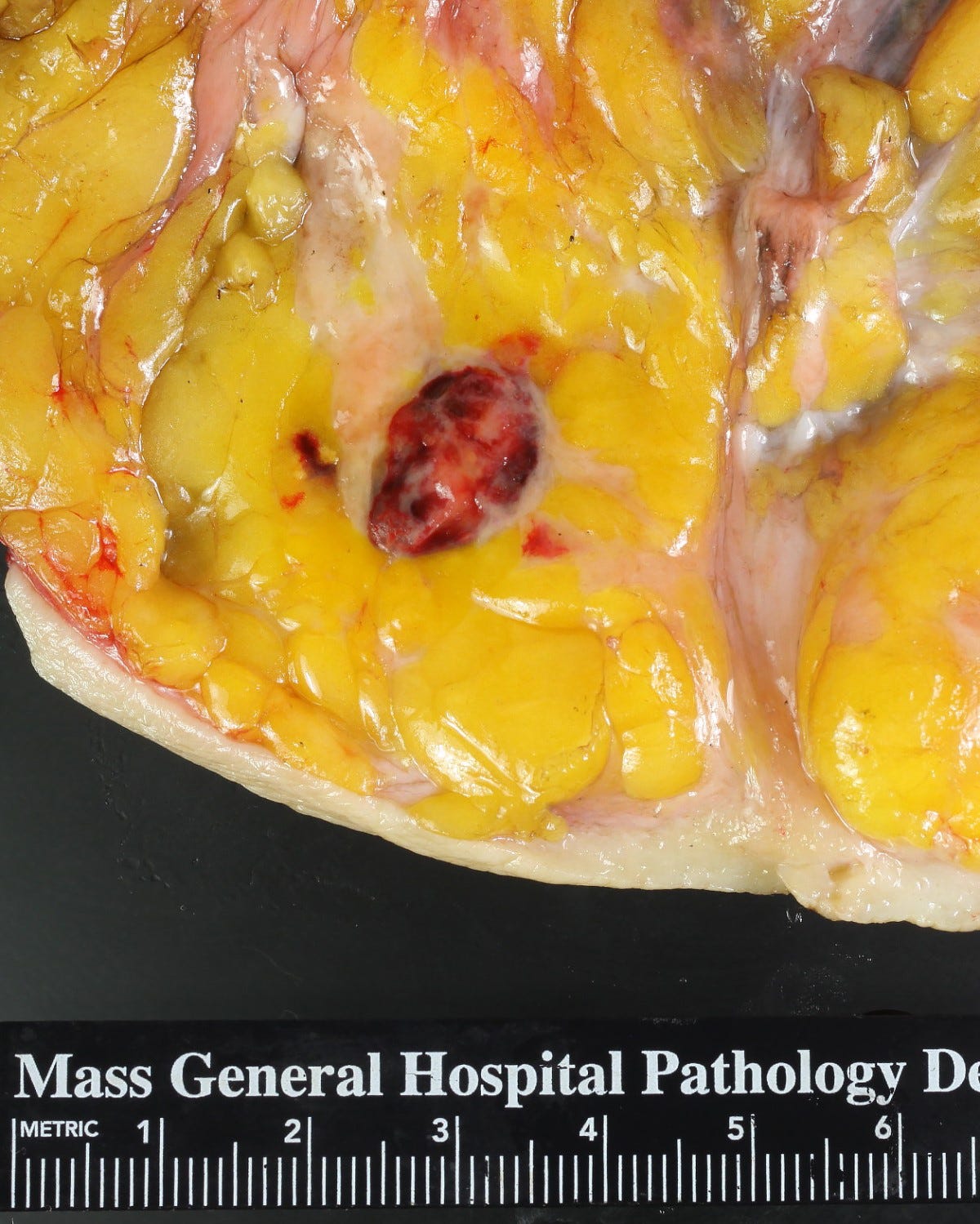
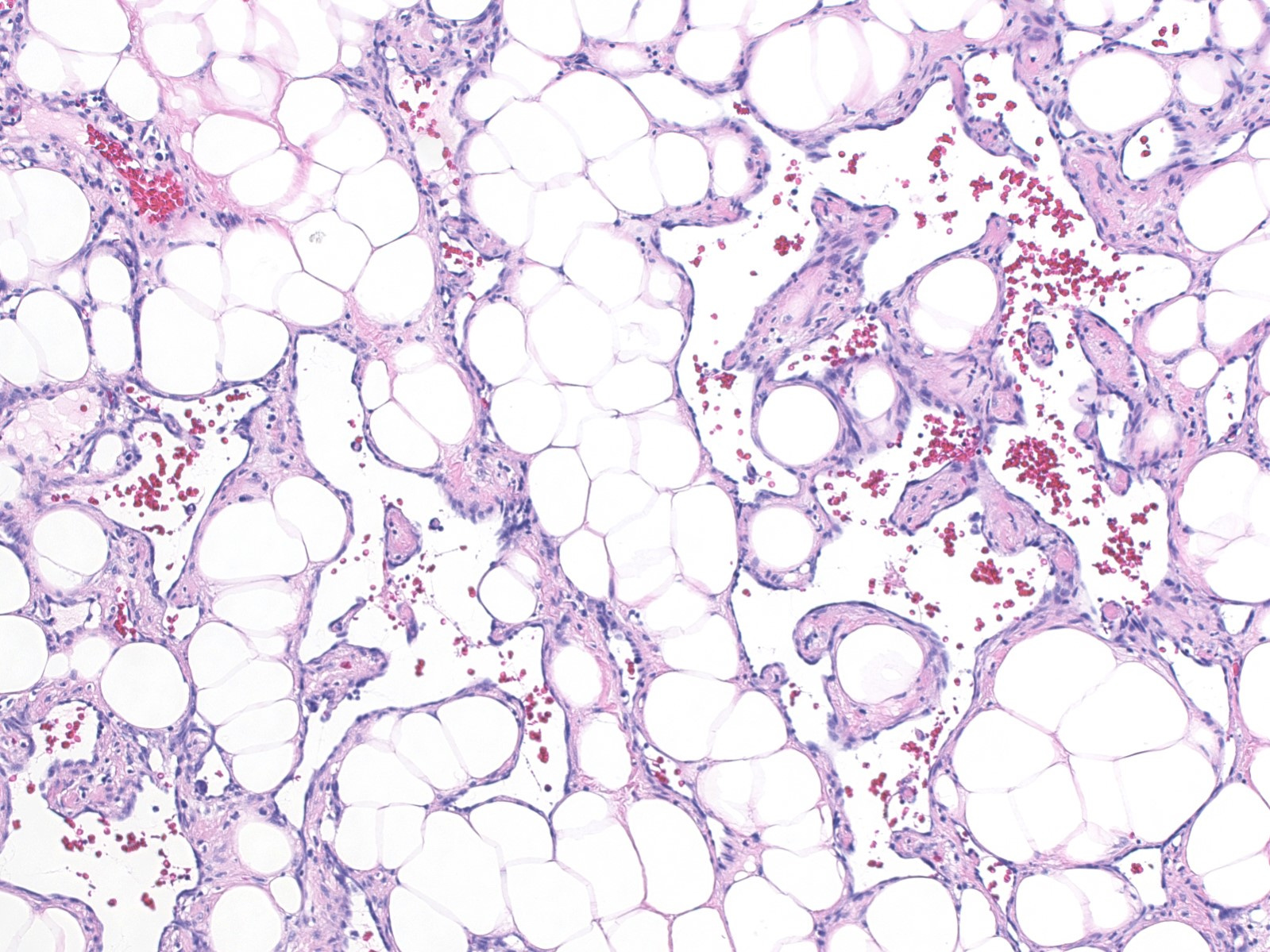
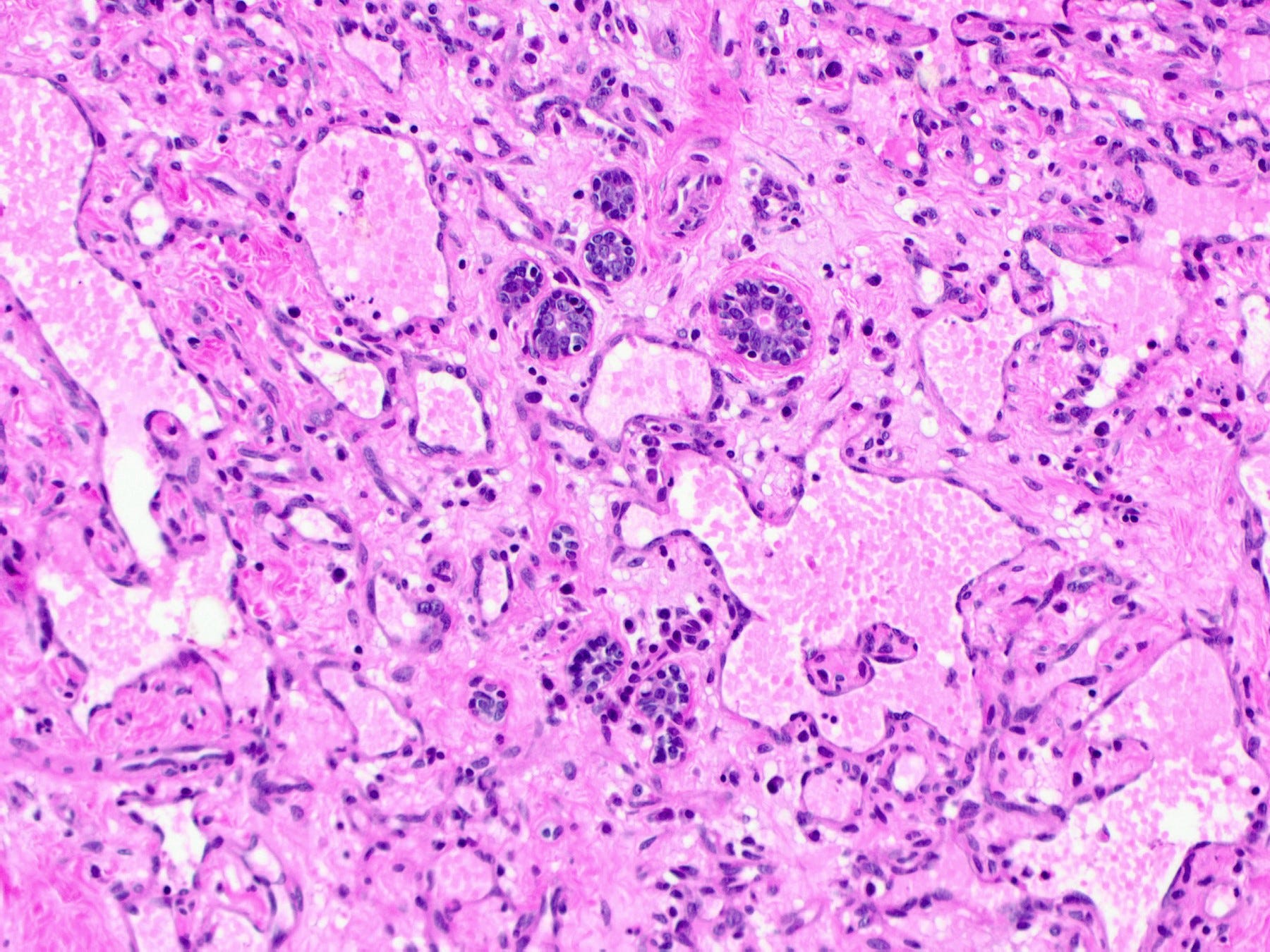
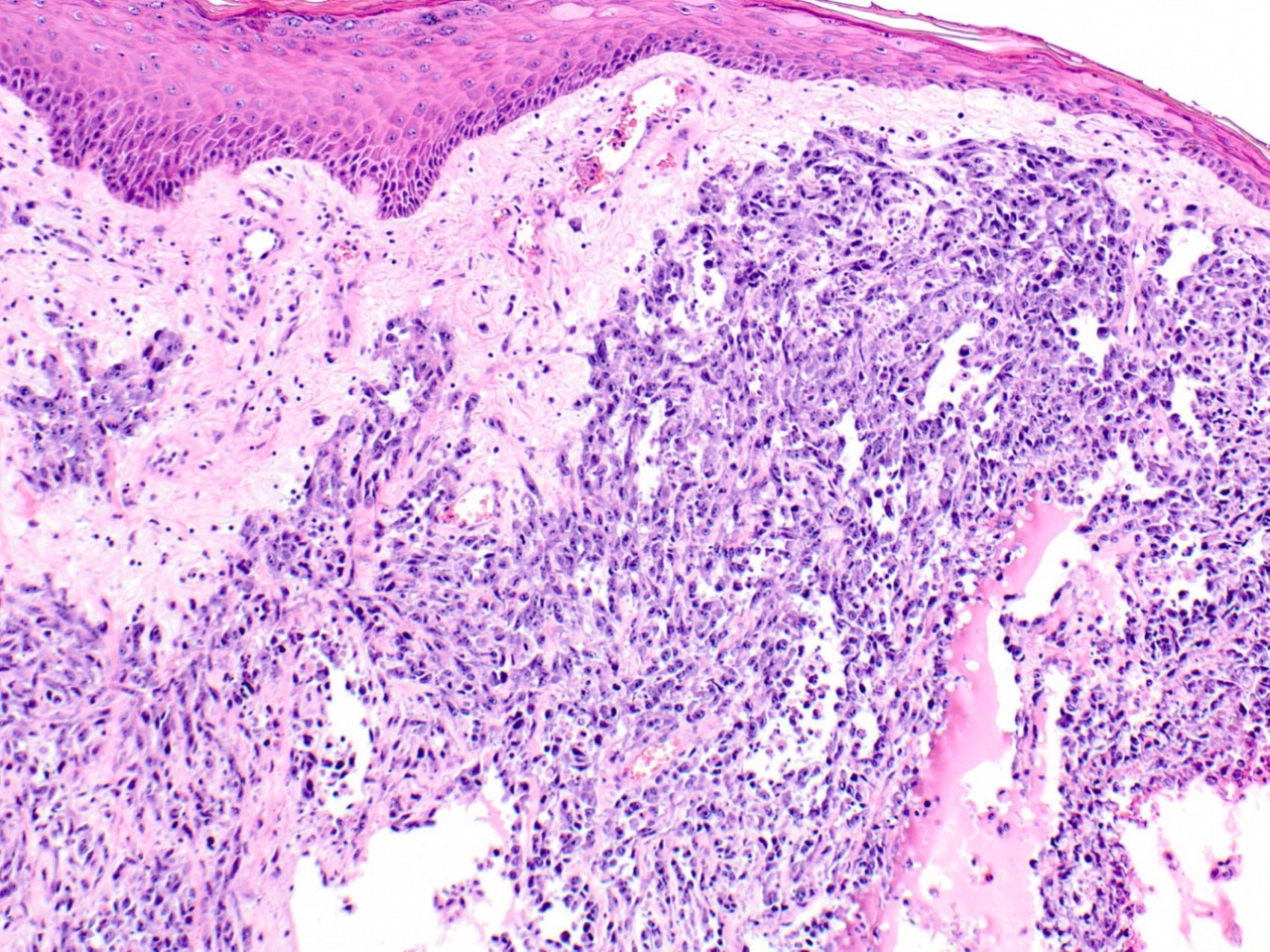
Epithelioid angiosarcoma of the breast
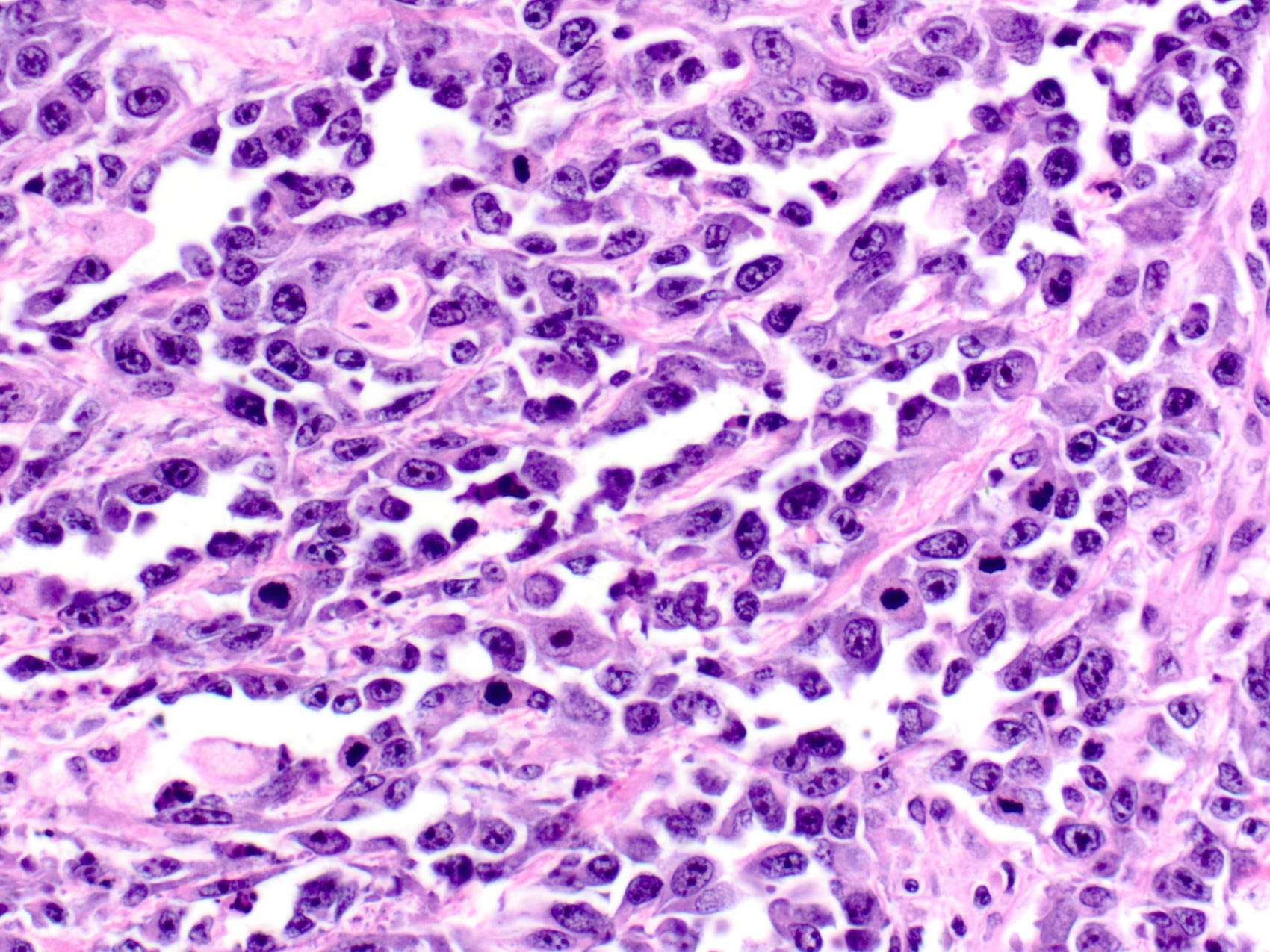
Epithelioid angiosarcoma is a rare and aggressive variant of breast angiosarcoma composed almost exclusively of epithelioid endothelial cells, i.e., cells that look epithelial but are not. They appear large, rounded or polygonal with abundant eosinophilic cytoplasm and prominent nucleoli.
Epithelioid angiosarcoma of the breast is similar to classic angiosarcoma because it may occur as a primary (de novo) tumor or secondary to radiation therapy with or without chronic lymphedema. No precursor lesion has been identified.
Epithelioid angiosarcoma differs from classic angiosarcoma of the breast in the following ways:
It arises from mesenchymal cells that display an epithelial morphology but are not epithelial.
It is more likely to present with early nodal and solid organ metastasis.
It tends to form solid sheets rather than well formed vascular channels.
It has numerous mitotic figures.
It commonly has necrosis in high grade tumors.
It may express cytokeratins and EMA (note that focal expression may also occur in high grade classic angiosarcomas).
It has a poorer prognosis with reported 5 year overall survival rates of 20 - 35%.
Epithelioid angiosarcomas may be confused with epithelial breast cancers although they express vascular markers and are typically triple negative. Rare reports of HER2+ expression may reflect technical artifacts.
The epithelioid morphology of some nonepithelial tumors, including epithelioid angiosarcoma, reflects phenotypic mimicry rather than true lineage transdifferentiation (i.e. its morphology appears epithelial but is actually not).
At other sites, genetic changes are associated with epithelioid features:
SMARCB1 (INI1) loss – seen in epithelioid sarcoma, malignant rhabdoid tumor of the kidney and other tumors.
TFE3 translocations – seen in alveolar soft part sarcomas.
However, genetic changes associated with epithelioid morphology have not yet been found in the breast.
Epithelioid angiosarcoma of the breast - microscopic images
Note: due to its rarity in the breast, some images are from other sites.
Breast implant associated anaplastic large cell lymphoma
Breast implant associated anaplastic large cell lymphoma, first recognized in 2017 by the World Health Organization, is a rare T cell lymphoma associated with breast implants. Its incidence varies by manufacturer from 1 per 2,000 to 30,000 women with textured breast implants.
It presents at a median 8 - 10 years after implant placement, most commonly in women in their mid 40’s to 50’s. These women often have a late developing seroma (fluid collection) at the implant site or a palpable mass, capsular contracture or a skin rash. Unlike primary breast carcinomas, they develop in the fibrous capsule surrounding the implant, not from the breast parenchyma.
Diagnosis requires a high index of suspicion. Ultrasound may detect an effusion or mass. MRI may also be useful. Fine needle aspiration of the seroma fluid is the preferred diagnostic procedure when a seroma is present; core biopsies are used for masses.
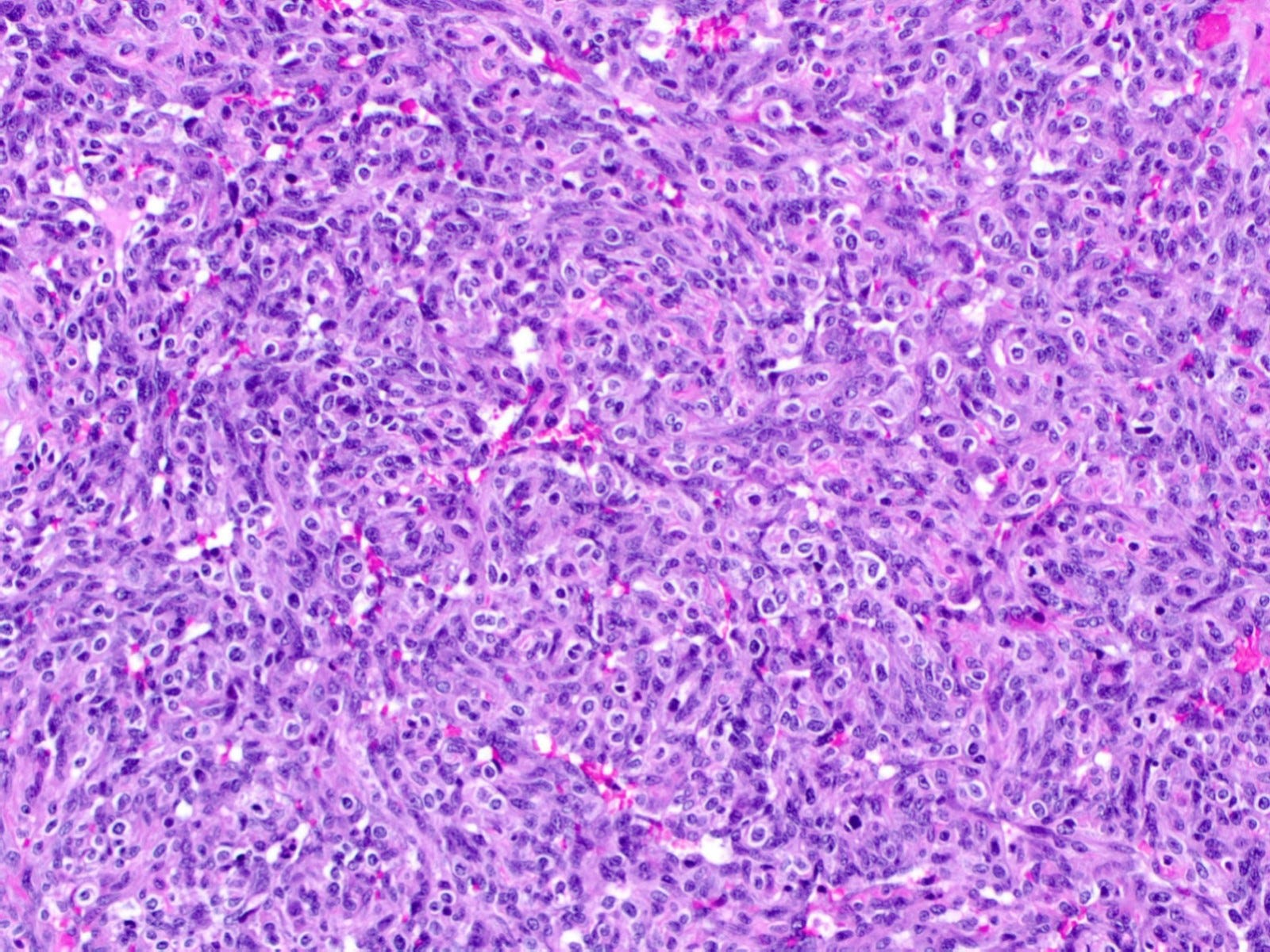
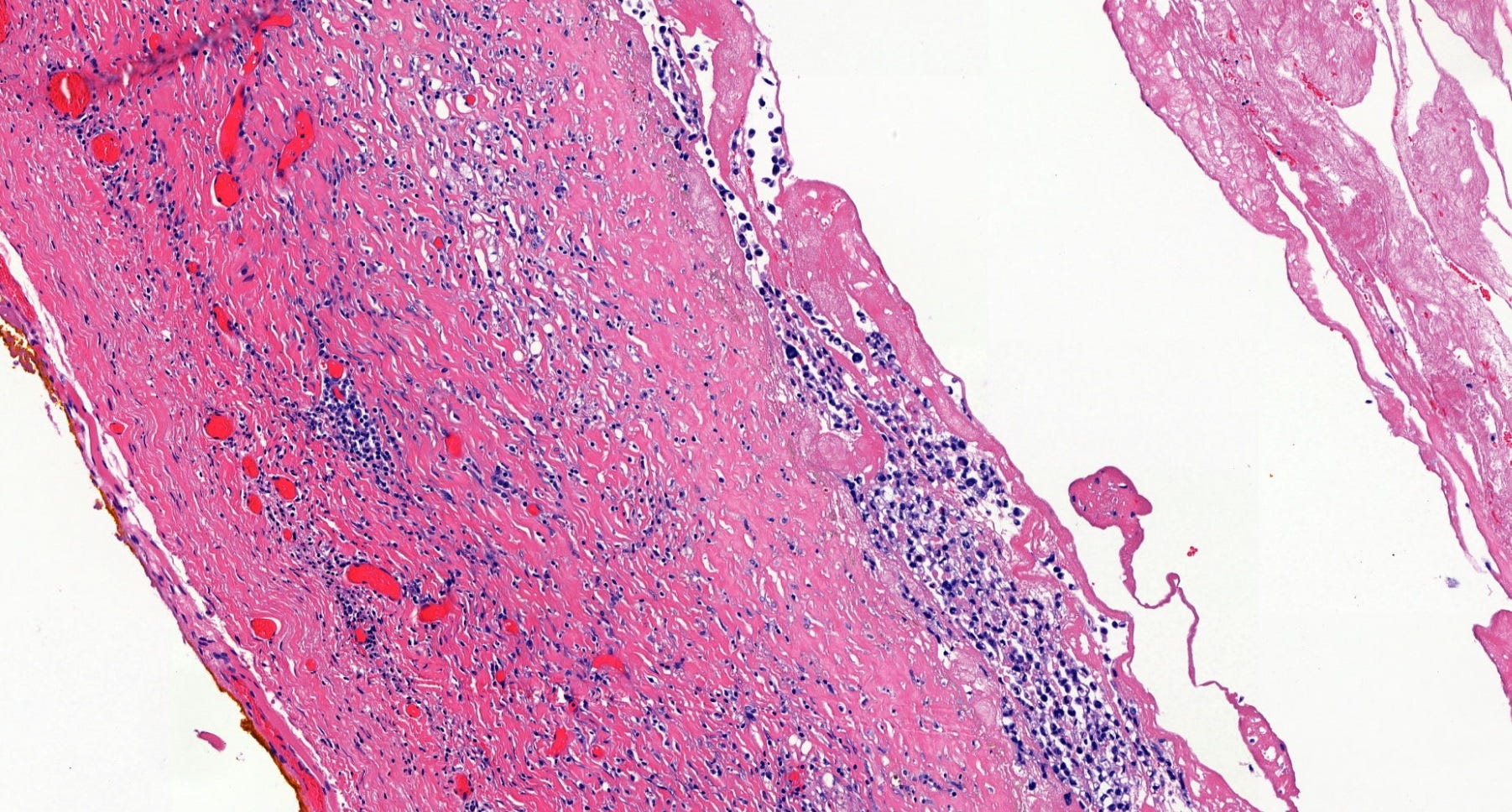
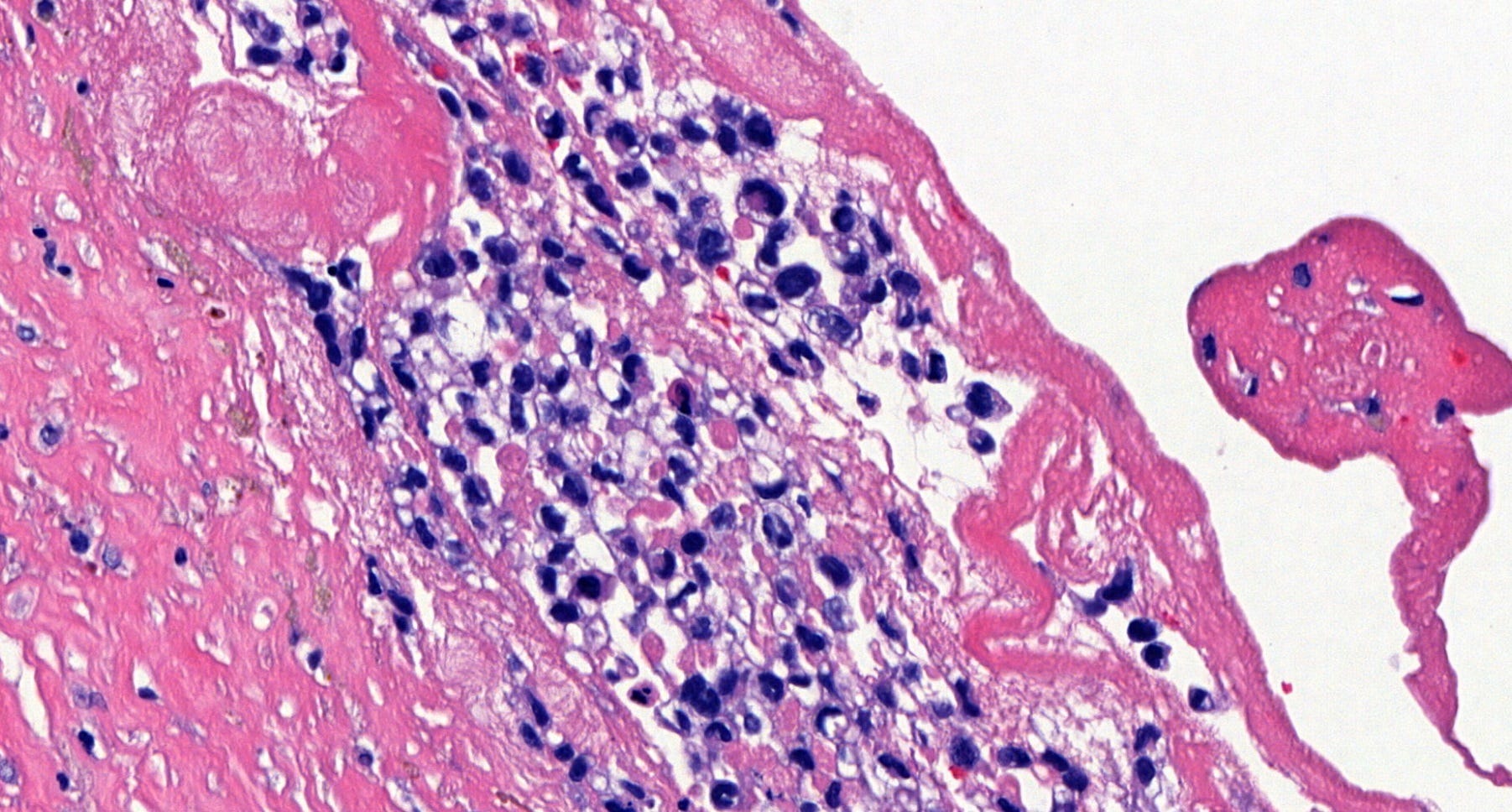
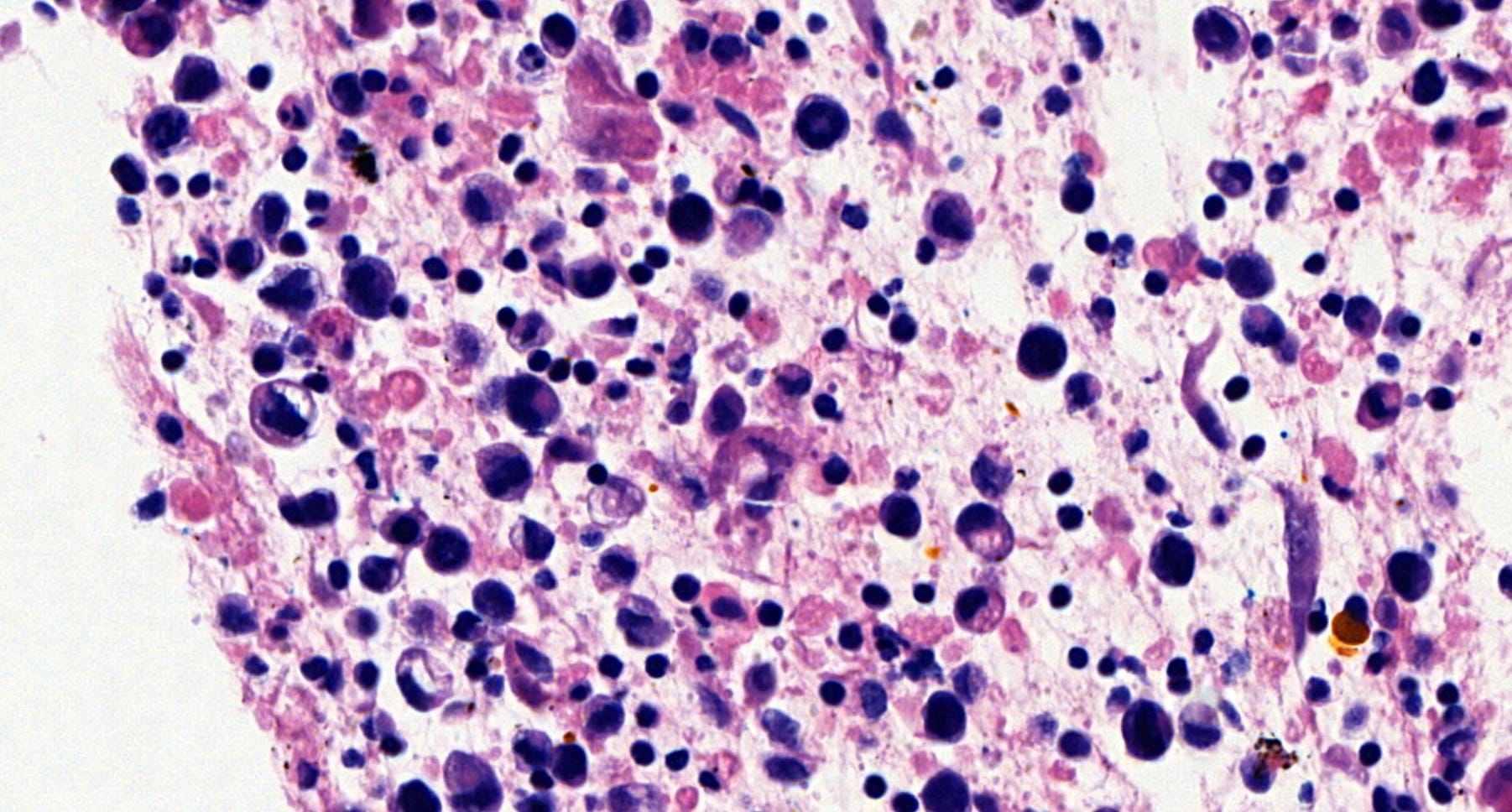
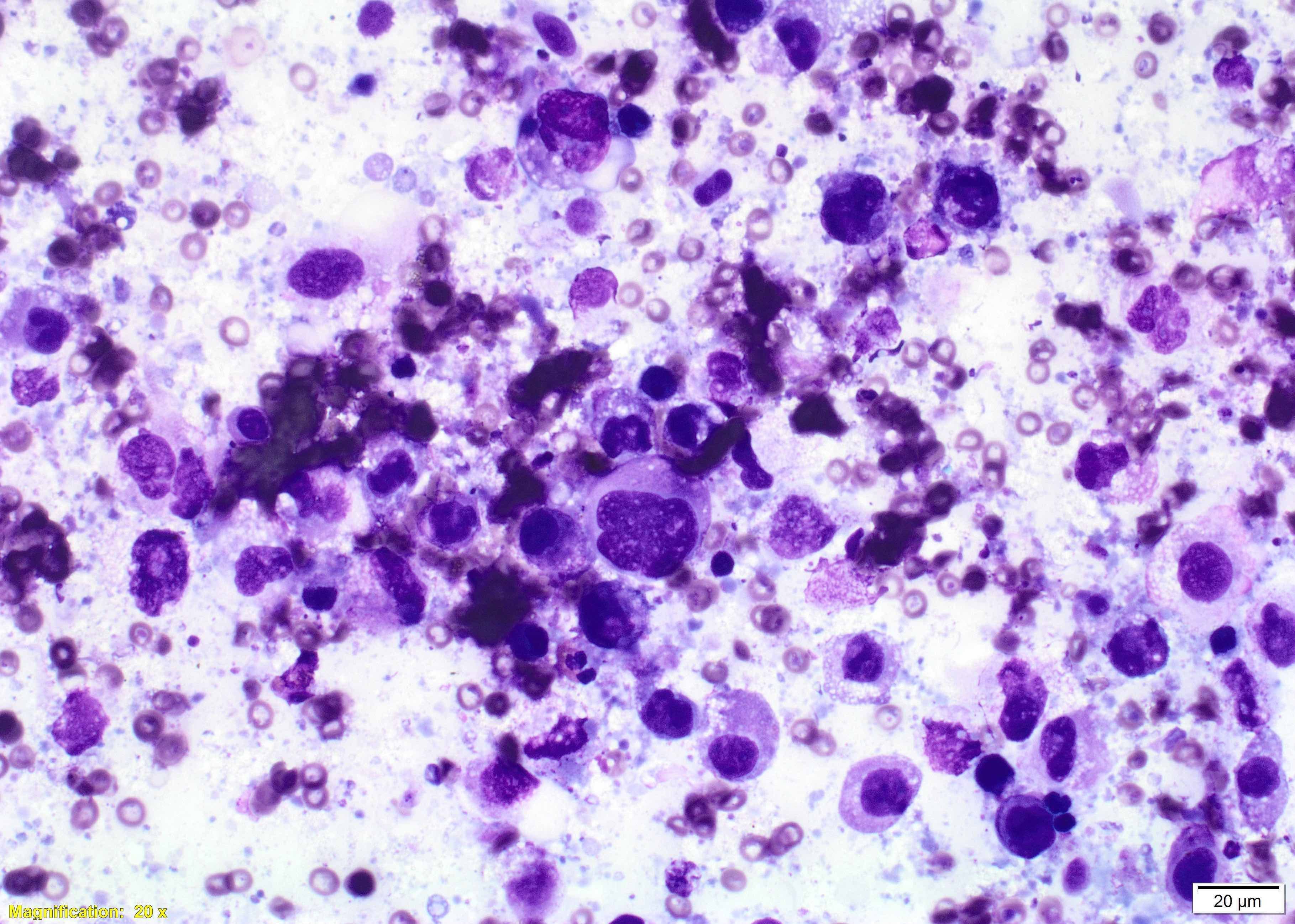
Microscopically, breast implant associated anaplastic large cell lymphoma is characterized by large pleomorphic lymphoid cells with abundant cytoplasm and horseshoe shaped nuclei. The malignant cells infiltrate the periprosthetic capsule and sometimes form masses with variable amounts of necrosis and inflammation. The tumor cells express CD30 and variably express T cell markers CD2, CD3, CD4 and CD43. They are negative for B cell markers. Unlike systemic anaplastic large cell lymphoma, which can be ALK positive or negative, this breast lymphoma is universally ALK negative, reflecting a distinct pathogenesis. Almost all cases are triple negative (i.e. negative for ER, PR and HER2), consistent with their lymphoid (not epithelial) origin
Treatment varies by stage. In early stage disease that is confined to the capsule (most cases), complete surgical excision of the implant and the entire surrounding capsule is usually curative. Axillary lymph node dissection is generally not indicated unless there is confirmed nodal involvement. Advanced or invasive disease (involving regional lymph nodes or distant organs) may require the addition of CHOP chemotherapy (cyclophosphamide, doxorubicin, vincristine and prednisone), targeted agents such as brentuximab vedotin (an anti-CD30 antibody drug conjugate) or possibly radiotherapy.
When diagnosed early, confined to the capsule and treated surgically, the prognosis is favorable with an overall survival rate of 90%. Prognosis worsens in advanced or systemic cases.
Breast implant associated anaplastic large cell lymphoma may arise due to chronic inflammation, biofilm formation and increased surface area of textured implants.
Chronic inflammation occurs because the rough, sandpaper-like surface of textured implants causes irritation and inflammation in the surrounding tissue, which stimulates the immune system. This may cause T cell proliferation and genetic instability, which may promote the malignant transformation of T cells.
Bacterial biofilms form due to the large surface area on textured surfaces. These biofilms also cause chronic inflammation and stimulate the immune system.
Breast implant associated anaplastic large cell lymphoma - microscopic images
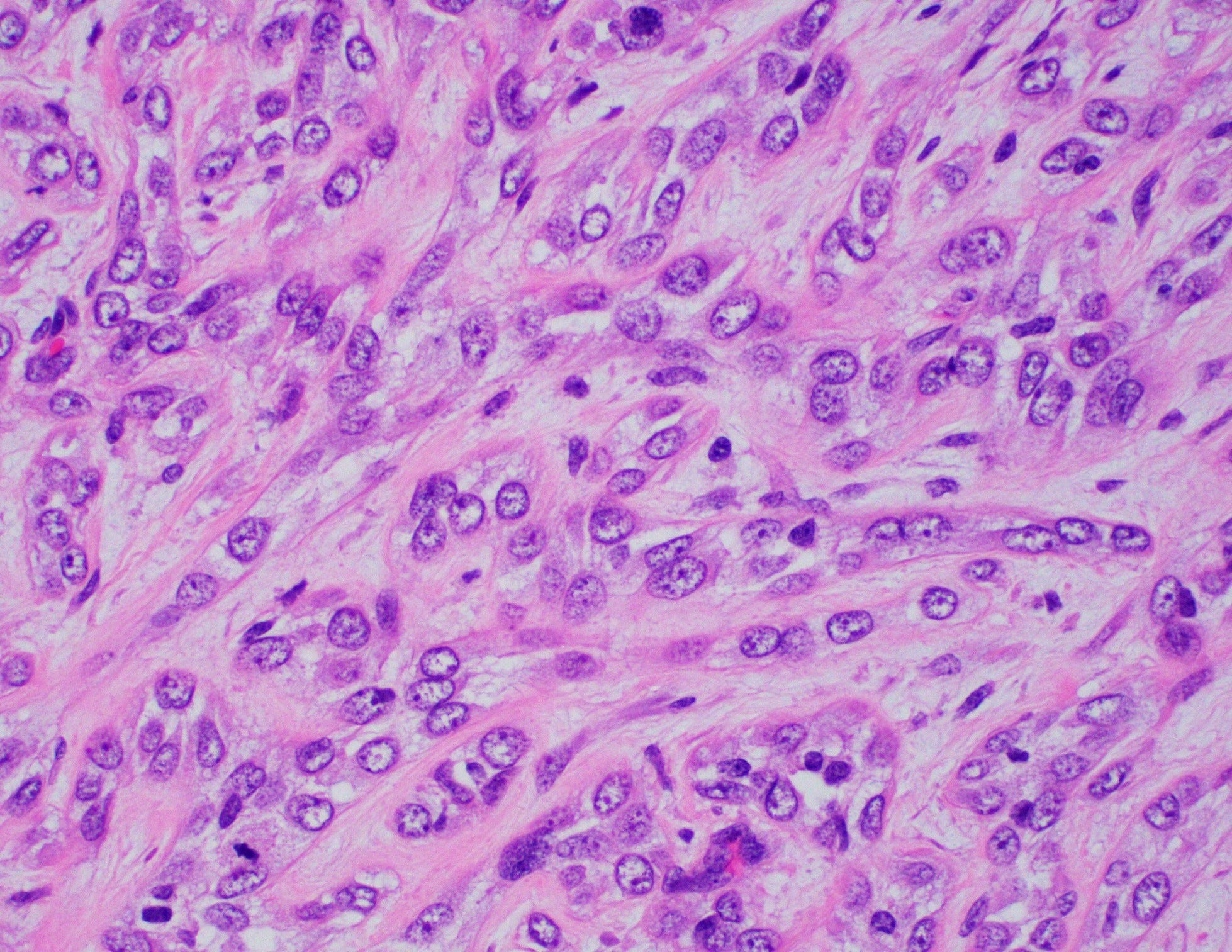
Myoepithelial carcinoma of the breast
Myoepithelial carcinoma of the breast is exceptionally rare (< 0.1% of all breast cancers). The mean age in the breast is 50 years, range 45 - 86 years. The preferred terminology is metaplastic carcinoma with myoepithelial differentiation.
It arises from the malignant transformation of myoepithelial cells, which are contractile cells between the basal lamina and luminal epithelial cells in the mammary ducts and lobules. These tumors can occur de novo or from the malignant progression of preexisting benign or atypical myoepithelial proliferations, such as adenomyoepithelioma or myoepitheliosis (proliferation of myoepithelial cells).
Due to the tumor’s rarity and lack of distinctive imaging characteristics, diagnosis is challenging. Diagnosis relies heavily on tissue biopsy, with core needle biopsy preferred over fine needle aspiration.
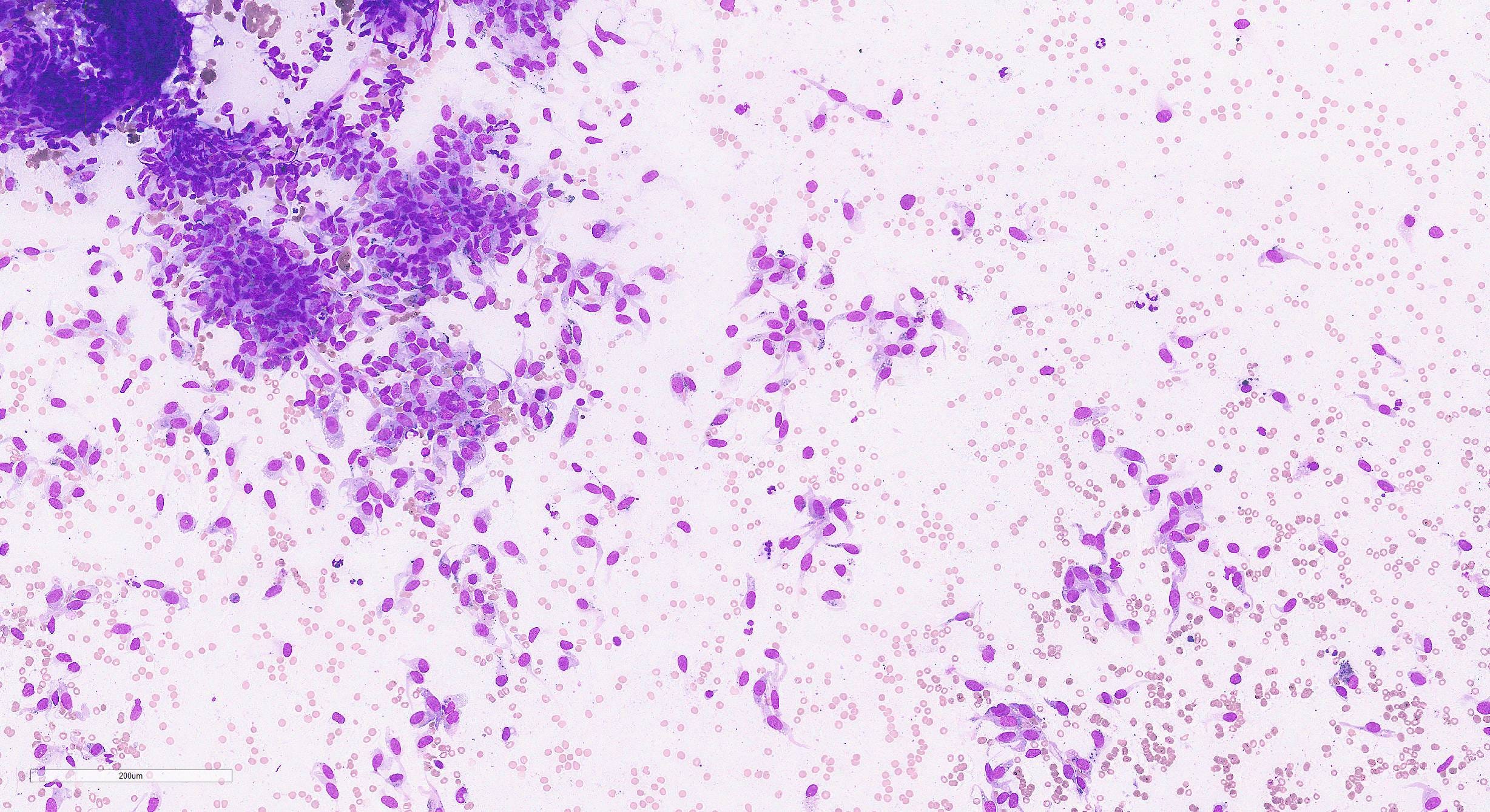
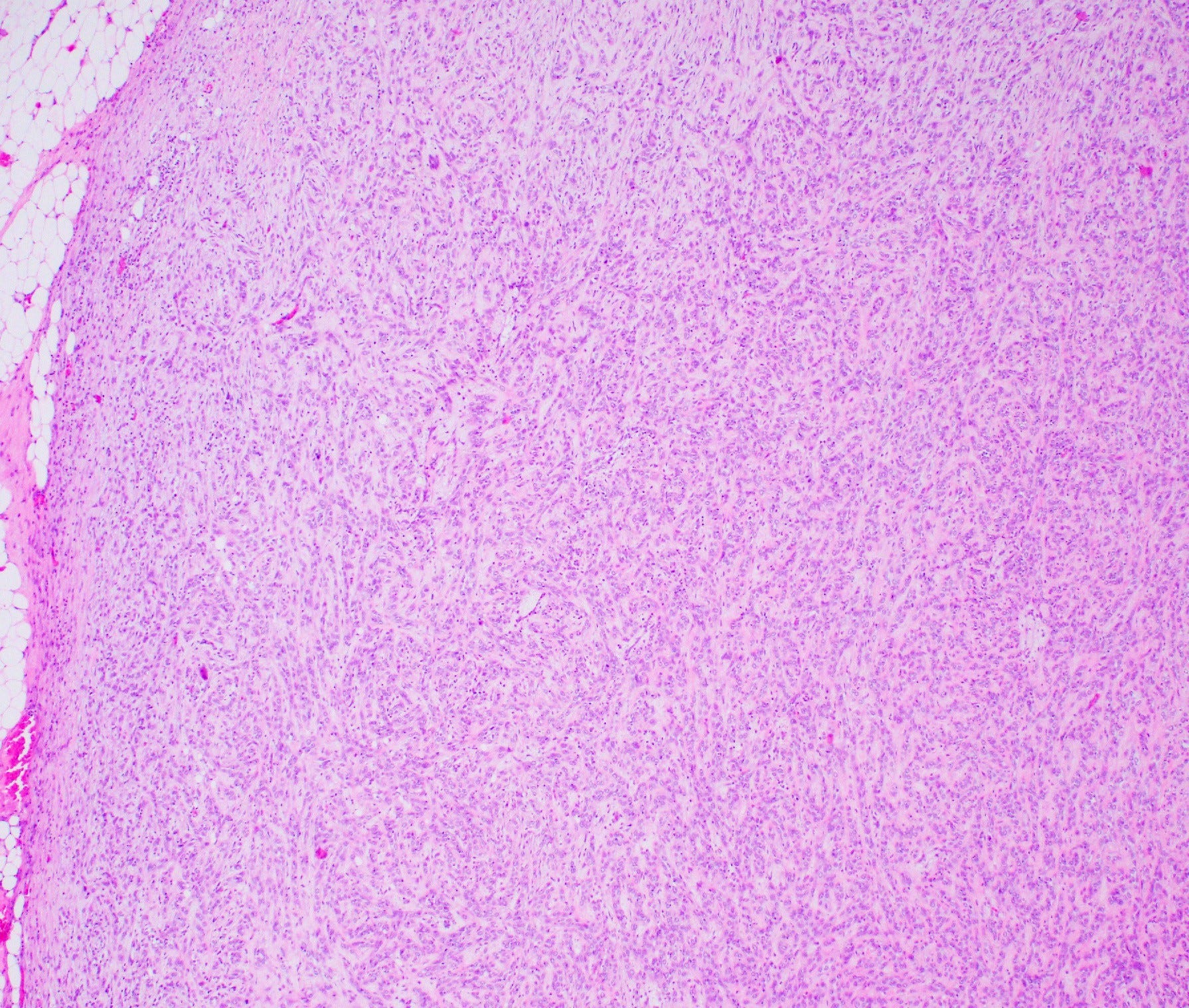
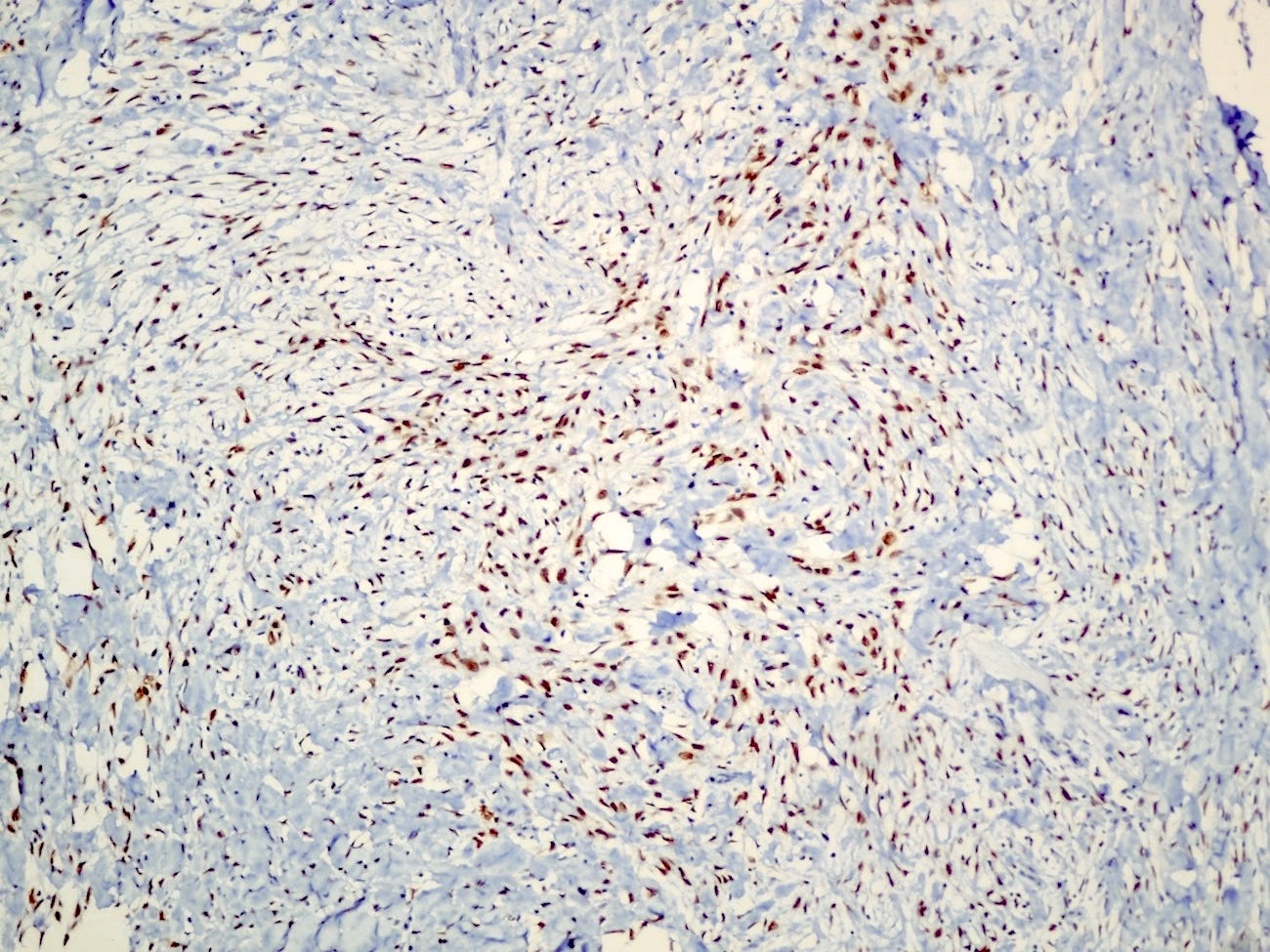
Histologically, myoepithelial carcinoma is composed of purely malignant myoepithelial cells, primarily spindle shaped to polygonal, possibly with cell clearing. They are arranged in solid, trabecular or reticular patterns. Tumor cells often exhibit nuclear atypia, increased mitotic figures and necrosis in high grade lesions. The stroma may range from sclerotic to myxoid. Mitotic figures are usually readily identifiable. Immunohistochemically, these tumors are triple negative (i.e., negative for ER, PR and HER2). They often express myoepithelial markers p63, CD10 and CK5/6 and EGFR, which helps distinguish them from other triple negative breast tumors.
Treatment is primarily surgical excision with negative margins, with individual determinations on the use of regional lymph node excision and radiotherapy. The role of chemotherapy is unclear.
Myoepithelial carcinoma is associated with an aggressive course, including recurrence and metastasis. Its 2 and 5 year survival is 88% and 55%, respectively. An increased tumor size is associated with a worse outcome.
Myoepithelial carcinoma originates from cells with dual epithelial and smooth muscle-like differentiation. In normal physiology, myoepithelial cells play essential roles in maintaining the structure of ductal units, supporting epithelial integrity and facilitating milk ejection through contractile function. Their transformation into malignancy likely involves genetic mutations and alterations in pathways regulating cell adhesion, apoptosis and proliferation.
The absence of hormone receptor expression and HER2 amplification suggests that these tumors diverge early from the typical luminal differentiation pathway of most breast cancers.
Myoepithelial carcinoma of the breast - microscopic images
The next essay will discuss neuroendocrine types of breast cancer (neuroendocrine tumors, neuroendocrine carcinoma (small cell and large cell types).
If you like these essays, please subscribe or share them with others.
Click here for the Index to Nat’s blog on Cancer and Medicine.
Follow me on LinkedIn, Threads and Instagram (@npernickmich) or Bluesky (@natpernick.bsky.social).
Follow our Curing Cancer Network through our Curing Cancer Newsletter, on LinkedIn or the CCN section of our PathologyOutlines.com blog. Each week, we post interesting cancer related images of malignancies with diagnoses, plus articles of interest. Please also read our CCN essays.
Latest cancer related documents:
American Code Against Cancer (how you can prevent cancer)
Email: Nat@PathologyOutlines.com (please note I cannot provide medical advice)
Follow my Substack Notes — subscribers are automatically notified.